
什么是单基因遗传病?
单基因遗传病是指由单个基因变异引起的疾病,代表性疾病包括:地中海贫血、遗传性耳聋、苯丙酮尿症、脊髓性肌萎缩症等。
我国是人口大国,也是出生缺陷高发国家,我国出生缺陷发生率在5.6%左右,每年新增出生缺陷数约90万例,其中22.2%的出生缺陷患儿是由单基因遗传病导致。
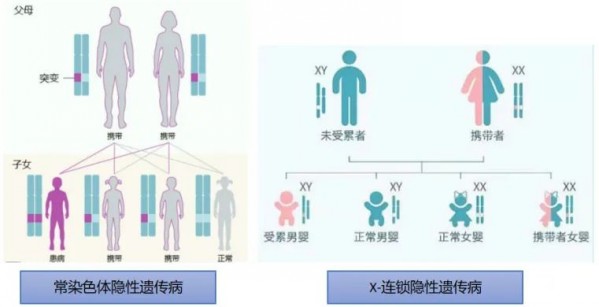
单基因遗传病多为隐性遗传,隐性遗传的单基因病携带者,虽自身无临床表现,但其缺陷基因会有50%的几率遗传给下一代。单基因遗传病可致死、致畸、致残,缺乏有效的治疗手段或治疗费用昂贵,基于此,单基因遗传病携带者的筛查显得尤为重要,通过孕期或孕早期的单基因遗传病携带者筛查可有效将出生缺陷防控端口前移,降低重症遗传病患儿出生。
遗传模式图
免费筛查活动介绍
单基因遗传病扩展性携带者筛查活动是由郑州大学第一附属医院遗传与产前诊断中心发起,20余家医疗机构共同参与,筛查常见11种单基因遗传病,本次招募为第3期,共80对名额。符合以下条件者可参与:
1、孕前或孕周≤13+6;
2、女方年龄18-40岁、男方年龄≥18岁;
3、受检者无遗传病家族史,未生育过单基因病患儿;
4、受检者非已知致病基因携带者;
5、受检者智力正常、无明显外观异常;
6、夫妇双方需同时采样,如已怀孕要求男方为胎儿生物学父亲,如未怀孕则要求男女双方非三代以内近亲;
7、受检者一年内未接收异体输血、器官移植或免疫治疗;
活动日期:
2023年11月10日-12月10日;
咨询地址:
门诊楼2楼妇产科诊区遗传咨询门诊;
咨询电话:
0377--61609318、15136677677 (陈医生)
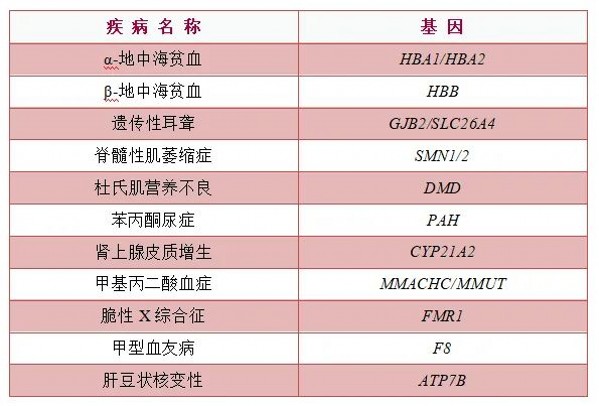
筛查病种
疾病简介:
地中海贫血,是由一种或几种珠蛋白肽链合成障碍引起的遗传性溶血性疾病。我国患者主要分布在海南、广西、广东等地区,发病率为10%~14%。患者的血红蛋白合成异常,导致红细胞携氧减少,从而出现器官缺氧。主要表现为不同程度的贫血、黄疸和脾肿大等症状。涉及珠蛋白的基因突变种类繁多,其中α和β珠蛋白生成障碍最常见,致病基因分别为HBA1、HBA2。
遗传性耳聋,是由于基因突变而导致的听力障碍,多由父母携带遗传性耳聋基因突变并遗传给后代所引起。目前研究显示,与耳聋相关的基因有200多个。最常见的致聋基因有GJB2、GJB3、SLC26A4、MT-RNR1等。正常人中约5%-6%的人至少携带一种耳聋基因突变。
脊髓性肌萎缩症,是一种以对称性肌无力和肌萎缩为特征的常染色体隐性遗传疾病,中国人群携带率高达1/50。主要致病基因为SMN1/2,患者的典型表现:肌无力、肌张力低、肌萎缩,正常站立、行走等运动功能受限,运动发育显著落后于正常儿童。严重者会在2岁前死于呼吸衰竭,是婴幼儿期遗传病头号杀手。且尚无法根治,患儿需终生用药控制病程发展。
杜氏肌营养不良症,是一种由DMD基因变异导致的进行性肌无力和肌肉萎缩性疾病。活产男婴中发病率约为1/3500,我国每年约有400-500例患儿出生,累计约7万人确诊,是世界上该病患者人数最多的国家之一。患者常以肌肉的进行性萎缩无力并伴有腓肠肌假性肥大为特征,多在3-5岁发病,若不治疗干预,通常在12岁前失去行走能力,轮椅为伴,大多在20至30岁期间死亡。
苯丙酮尿症,是一种由PAH基因变异导致的常染色体隐性遗传代谢病,使人体内苯丙氨酸代谢途径中的酶缺陷,导致苯丙氨酸(人体必需的一种氨基酸)代谢异常,使苯丙氨酸不能转变成酪氨酸,导致苯丙氨酸及其代谢产物,从尿中大量排出。升高的苯丙氨酸会通过血脑屏障,损伤人体大脑神经元,进而对智力发育造成不可逆的损害。
先天性肾上腺皮质增生症,为CYP21A2基因变异引起的常染色体隐性遗传性病,导致肾上腺类固醇激素生物合成过程某种酶(多为21-羟化酶)的先天性缺陷。我国于2007年开始新生儿筛查,患病率为1/12 200。患者临床表型为肾上腺皮质功能减退、电解质紊乱、性腺发育异常、身材矮小以及青春期和成年期代谢综合征的风险增加等,严重时可危及生命。
甲基丙二酸血症,是一种常染色体隐性遗传病,是由于甲基丙二酰辅酶A变位酶自身缺陷或者其辅酶钴胺素代谢缺陷所致甲基丙二酸等代谢物异常蓄积。致病基因为MMACHC/MMUT。临床表现复杂多样,如不能及时发现和治疗,可导致多脏器损害,以脑损害为主,还有一些患者表现为心血管、肾、肺、眼、骨髓及皮肤损害。
脆性X综合征,是一种以轻度至中度智力残疾为特征的遗传性疾病,95%以上是由FMR1基因变异引起。典型患者多见于男性,患者智力只有正常人的 40%,且会随着年龄的增长逐渐降低。面部特征包括长而狭窄的脸、大耳朵、前额突出,男性患者会在青春期表现出大睾丸。约有三分之一的患者患有自闭症,10%-20%的患者有癫痫发作史,一些患者还有多动症、攻击性行为。女性患者与男性患者表现类似,但程度较轻。
血友病,是指因凝血因子Ⅷ(血友病A)、因子Ⅸ(血友病B)或因子Ⅺ(血友病C)缺乏所导致的遗传性出血性疾病。男性患病,女性一般为携带者;患者表现为轻微创伤后出血倾向或“自发性”出血,比如皮肤总会出现瘀斑、鼻腔出血、牙龈出血、关节出血等,凝血功能异常。甲型血友病,又称血友病A,致病基因为F8基因,呈X连锁隐性遗传,男性患者子女均不患病(其妻子为野生型),但其可将致病基因传递给女儿。
肝豆状核变性,又称Wilson病,是因铜转运ATP酶β(ATP7B)基因突变而导致的铜代谢障碍性疾病。该病临床表现复杂,主要为肝脏和神经系统病变,多见于儿童、青少年。因受累器官和程度不同而异,主要表现为肝脏和/或神经系统受累。此外,还可出现眼部异常、溶血、肾脏损伤、骨关节异常等多种临床表现。
|
